scDblFinder
Pierre-Luc Germain
University and ETH ZürichSource:
vignettes/scDblFinder.Rmd
scDblFinder.RmdAbstract
An introduction to the scDblFinder method for fast and comprehensive doublet identification in single-cell data.
scDblFinder
The scDblFinder method combines the strengths of various
doublet detection approaches, training an iterative classifier on the
neighborhood of real cells and artificial doublets.
scDblFinder() has two main modes of operation:
cluster-based or not. Both perform quite well (see Germain et al.,
2021). In general, we recommend the cluster-based approach in
datasets with a very clear cluster structure, and the random approach in
more complex datasets.
Installation
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("scDblFinder")
# or, to get that latest developments:
BiocManager::install("plger/scDblFinder")Usage
The input of scDblFinder is an object sce
of class SingleCellExperiment
(empty drops having already been removed) containing at least the counts
(assay ‘counts’). Alternatively, a simple count matrix can also be
provided.
Given an SCE object, scDblFinder (using the random
approach) can be launched as follows :
set.seed(123)
suppressPackageStartupMessages(library(scDblFinder))
# we create a dummy dataset; since it's small we set a higher doublet rate
sce <- mockDoubletSCE(dbl.rate=0.1, ngenes=300 )
# we run scDblFinder (providing the unusually high doublet rate)
sce <- scDblFinder(sce, dbr=0.1)## Creating ~1500 artificial doublets...## Dimensional reduction## Evaluating kNN...## Training model...## iter=0, 37 cells excluded from training.## iter=1, 26 cells excluded from training.## iter=2, 28 cells excluded from training.## Threshold found:0.382## 28 (5.3%) doublets calledFor 10x data, it is usually safe to leave the dbr empty,
and it will be automatically estimated. (If using a chip other than the
standard 10X, you might have to adjust it or the related
dbr.per1k argument.
scDblFinder will add a number of columns to the colData
of sce prefixed with ‘scDblFinder’, the most important of
which are:
-
sce$scDblFinder.score: the final doublet score -
sce$scDblFinder.class: the classification (doublet or singlet)
We can compare the calls with the truth in this toy example:
table(truth=sce$type, call=sce$scDblFinder.class)## call
## truth singlet doublet
## singlet 496 4
## doublet 0 24Since most of the time the truth is not known, a good, simple diagnostic is the distribution of doublet score:
hist(sce$scDblFinder.score)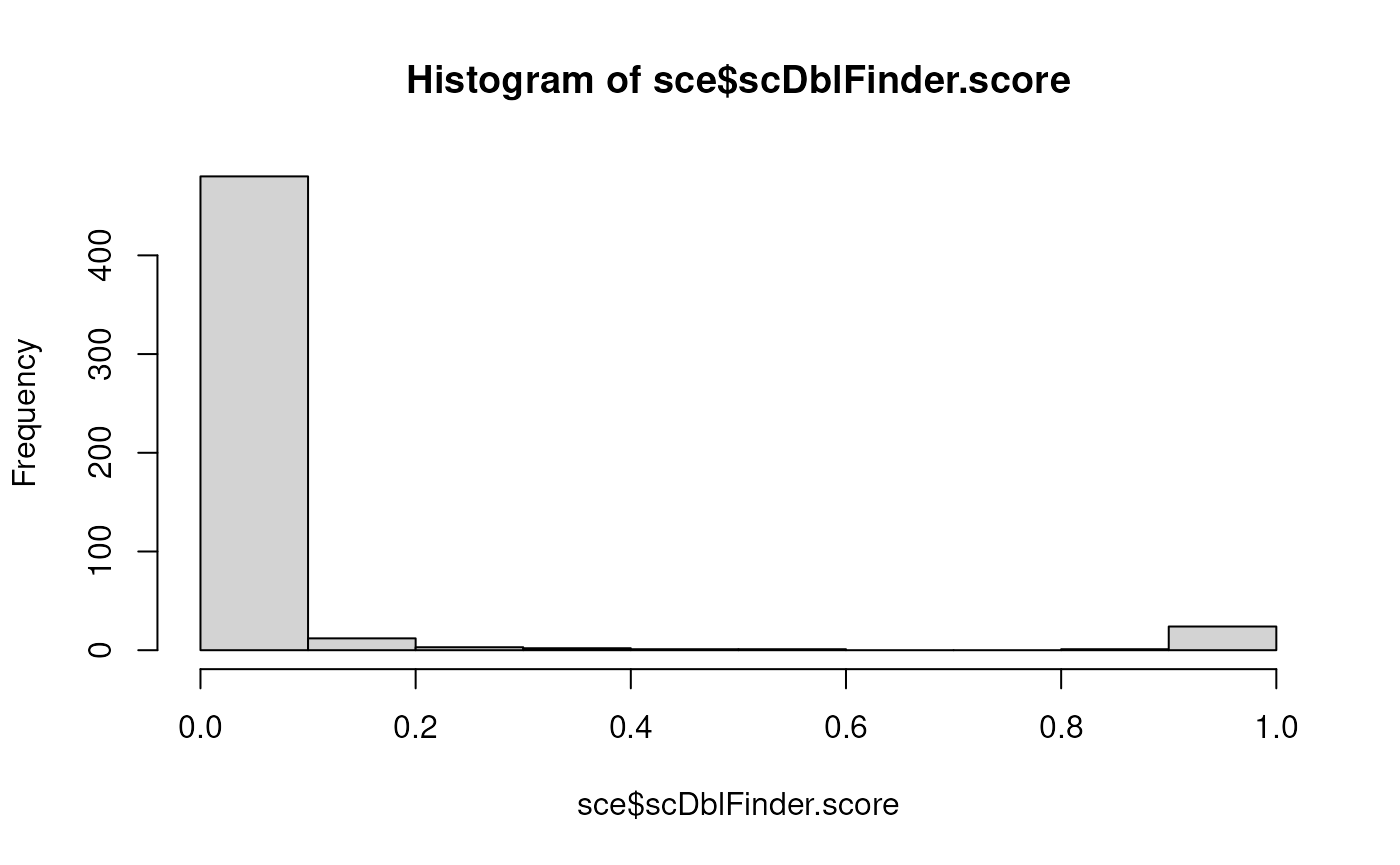 A bimodal distribution, with most values very close to 0, a few close to
1, and very little in-between is the sign that scDblFinder is able to do
its job.
A bimodal distribution, with most values very close to 0, a few close to
1, and very little in-between is the sign that scDblFinder is able to do
its job.
Cluster-based approach
To use the cluster-based approach, one simply needs to additionally
provide the clusters argument:
sce <- scDblFinder(sce, clusters="cluster")## 2 clusters## Creating ~1500 artificial doublets...## Dimensional reduction## Evaluating kNN...## Training model...## iter=0, 24 cells excluded from training.## iter=1, 24 cells excluded from training.## iter=2, 24 cells excluded from training.## Threshold found:0.998## 24 (4.6%) doublets called
table(truth=sce$type, call=sce$scDblFinder.class)## call
## truth singlet doublet
## singlet 500 0
## doublet 0 24The clusters argument can be either a vector of cluster
labels for each column of sce, a colData column of
sce containing such labels, or TRUE. If
clusters=TRUE, the fast clustering approach (see
?fastcluster) will be employed. If normalized expression
(assay ‘logcounts’) and/or PCA (reducedDim ‘PCA’) are already present in
the object, these will be used for the clustering step.
Multiple samples
If you have multiple samples (understood as different cell captures),
then it is preferable to look for doublets separately for each sample
(for multiplexed samples with cell hashes, this means for each batch).
You can do this by simply providing a vector of the sample ids to the
samples parameter of scDblFinder or, if these
are stored in a column of colData, the name of the column.
In this case, you might also consider multithreading it using the
BPPARAM parameter (assuming you’ve got enough RAM!). For
example:
library(BiocParallel)
sce <- scDblFinder(sce, samples="sample_id", BPPARAM=MulticoreParam(3))
table(sce$scDblFinder.class)Note that if you are running multiple samples using the cluster-based
approach (see below), clustering will be performed sample-wise. While
this is typically not an issue for doublet identification, it means that
the cluster labels (and putative origins of doublets) won’t match
between samples. If you are interested in these, it is preferable to
first cluster (for example using
sce$cluster <- fastcluster(sce)) and then provide the
clusters to scDblFinder, which will ensure concordant
labels across samples.
Of note, if you have very large differences in number of cells between samples the scores will not be directly comparable. We are working on improving this, but in the meantime it would be preferable to stratify similar samples and threshold the sets separately.
Description of the method
Wrapped in the scDblFinder function are the following
steps:
Splitting captures
Doublets can only arise within a given sample or capture, and for
this reason are better sought independently for each sample, which also
speeds up the analysis. If the samples argument is given,
scDblFinder will use it to split the cells into
samples/captures, and process each of them in parallel if the
BPPARAM argument is given. Depending on the
multiSampleMode argument, the classifier can be trained
globally, with thresholds optimized on a per-sample basis; however we
did not see an improvement in doing so, and therefore by default each
sample is treated separately to maximize robustness to technical
differences.
If your samples are multiplexed, i.e. the different samples are mixed in different batches, then the batches should be what you provide to this argument.
Reducing and clustering the data
The analysis can be considerably sped up, at little if any cost in
accuracy, by reducing the dataset to only the top expressed genes
(controlled by the nfeatures argument).
Then, depending on the clusters argument, an eventual
PCA and clustering (using the internal fastcluster
function) will be performed. The rationale for the cluster-based
approach is that homotypic doublets are nearly impossible to distinguish
on the basis of their transcriptome, and therefore that creating that
kind of doublets is a waste of computational resources that can moreover
mislead the classifier into flagging singlets. An alternative approach,
however, is to generate doublets randomly (setting clusters
to FALSE or NULL), and use the iterative approach (see below) to exclude
also unidentifiable artificial doublets from the training.
Generating artificial doublets
Depending on the clusters and propRandom
arguments, artificial doublets will be generated by combining random
cells and/or pairs of non-identical clusters (this can be performed
manually using the getArtificialDoublets function). A
proportion of the doublets will simply use the sum of counts of the
composing cells, while the rest will undergo a library size adjustment
and poisson resampling.
Examining the k-nearest neighbors (kNN) of each cell
A new PCA is performed on the combination of real and artificial
cells, from which a kNN network is generated. Using this kNN, a number
of parameters are gathered for each cell, such as the proportion of
doublets (i.e. artificial doublets or known doublets provided through
the knownDoublets argument, if given) among the KNN, ratio
of the distances to the nearest doublet and nearest non-doublet, etc.
Several of this features are reported in the output with the
‘scDblFinder.’ prefix, e.g.:
-
distanceToNearest: distance to the nearest cell (real or artificial) -
ratio: the proportion of the KNN that are doublets. (If more than one value ofkis given, the various ratios will be used during classification and will be reported) -
weighted: the proportion of the KNN that are doublets, weighted by their distance (useful for isolated cells)
Training a classifier
Unless the score argument is set to ‘weighted’ or
‘ratio’ (in which case the aforementioned ratio is directly used as a
doublet score), scDblFinder then uses gradient boosted
trees trained on the kNN-derived properties along with a few additional
features (e.g. library size, number of non-zero features, and an
estimate of the difficultly of detecting artificial doublets in the
cell’s neighborhood, a variant of the cxds score from the
scds,
etc.) to distinguish doublets (either artificial or given) from other
cells, and assigns a score on this basis.
One problem of using a classifier for this task is that some of the
real cells (the actual doublets) are mislabeled as singlet, so to speak.
scDblFinder therefore iteratively retrains the classifier,
each time excluding from the training the (real) cells called as
doublets in the previous step (as well as unidentifiable artificial
doublets). The number of steps being controlled by the iter
parameter (in our experience, 2 or 3 is optimal).
This score is available in the output, in the
scDblFinder.score colData column, and can be interpreted as
a probability. If the data is multi-sample, a single model is trained
for all samples.
Thresholding
Rather than thresholding on some arbitrary cutoff of the score,
scDblFinder uses the expected number of doublets in
combination to the misclassification rate to establish a threshold.
Unless it is manually given through the dbr argument, the
expected doublet rate is first estimated (see below). If samples were
specified, and if the dbr is automatically calculated,
thresholding is performed separately across samples.
Thresholding then tries to simultaneously minimize: 1) the
classification error (in terms of the proportion of known doublets below
the threshold) and 2) the deviation from the expected number of doublets
among real cells (as a ratio of the total number of expected doublets
within the range determined by dbr.sd, and adjusted for
homotypic doublets). This means that, if you have no idea about the
doublet rate, setting dbr.sd=1 will make the threshold
depend entirely on the misclassification rate.
Doublet origins and enrichments
If artificial doublets are generated between clusters, it is
sometimes possible to call the most likely origin (in terms of the
combination of clusters) of a given putative real doublet. We observed
that at least one of the two composing cell is typically recognized, but
that both are seldom correctly recognized, owing to the sometimes small
relative contribution of one of the two original cells. This information
is provided through the scDblFinder.mostLikelyOrigin column
of the output (and the scDblFinder.originAmbiguous column
indicates whether this origin is ambiguous or rather clear). This, in
turn, allows us to identify enrichment over expectation for specific
kinds of doublets. Some statistics on each combination of clusters are
saved in metadata(sce)$scDblFinder.stats, and the
plotDoubletMap function can be used to visualize
enrichments. In addition, two frameworks are offered for testing the
significance of enrichments:
- The
clusterStickinessfunction tests whether each cluster forms more doublet than would be expected given its abundance, by default using a single quasi-binomial model fitted across all doublet types. - The
doubletPairwiseEnrichmentfunction separately tests whether each specific doublet type (i.e. combination of clusters) is more abundant than expected, by default using a poisson model.
Some important parameters
scDblFinder has a fair number of parameters governing
the preprocessing, generation of doublets, classification, etc. (see
?scDblFinder). Here we describe just a few of the most
important ones.
Expected proportion of doublets
The expected proportion of doublets has no impact on the density of
artificial doublets in the neighborhood, but impacts the classifier’s
score and, especially, where the cutoff will be placed. It is specified
through the dbr parameter, as well as
dbr.per1k (which specifies, if dbr is omitted,
the rate per thousands cells from which to estimate it). In addition,
the dbr.sd parameter specifies a +/- range around
dbr within which the deviation from dbr will
be considered null.
For most platforms, the more cells you capture the higher the chance
of creating a doublet. For standard 10X data, the 10X documentation
indicates a doublet rate of roughly 0.8% per 1000 cells captured, which
is the default value of dbr.per1k. This means that unless
dbr is manually set, with 5000 cells, (0.008*5)*5000 = 200
doublets are expected, and the default expected doublet rate will be set
to this value (with a default standard deviation of 0.015). Note however
that different protocols may vary in the expected proportion of
doublets. For example, the high-throughput (HT) 10X kit has an expected
doublet rate of half the standard, i.e. 0.4% per 1000 cells, so if using
that kit, set dbr.per1k=0.004.
Also note that strictly speaking, the proportion of doublets depends more on the number of cells inputted than that recovered. If your recovery rate was lower than expected, you might observe a higher doublet rate (see the too-many doublets section below).
The impact of the expected doublet rate on the thresholding will
depend on how hard the classification task is: if it is easy, the called
doublets will not depend much on the expected rate. If you are
unsure about the doublet rate, you might consider increasing
dbr.sd: with a high value (e.g. 1), the
thresholding will be entirely based on the misclassification error
(without any assumption about an expected doublet rate).
Number of artificial doublets
The number of artificial doublets can be set through the
artificialDoublets parameter. Using more artificial
doublets leads to a better sampling of the possible mixtures of cells,
but increases memory and runtime, and can skew the scores, in extreme
cases leading to difficulties in setting a threshold for being called as
a doublet (see this issue for
a discussion).
By default, scDblFinder will generate roughly as many
artificial doublets as there are cells, which is usually appropriate.
However, for very small datasets this could represent an undersampling
of the mixing space and hence lead to lower detection accuracy. For this
reason, a hard minimum number of artificial doublet is set. This will
tend to improve accuracy for small datasets, but the scores will be
skewed towards 1, possibly making a separation difficult. If you are in
such a situation and your histogram of scores does not show a
bimodality, consider manually setting the
artificialDoublets parameter to something closer to your
actual number of cells.
Frequently-asked questions
I’m getting way too many doublets called - what’s going on?
Then you most likely have a wrong doublet rate. If you did not
provide it (dbr argument), the doublet rate will be
calculated automatically using expected doublet rates from 10x, meaning
that the more cells captured, the higher the doublet rates. If you have
reasons to think that this is not applicable to your data, set the
dbr manually.
The most common cause for an unexpectedly large proportion of
doublets is if you have a multi-sample dataset and did not split by
samples. scDblFinder will think that the data is a single
capture with loads of cells, and hence with a very high doublet rate.
Splitting by sample should solve the issue.
Also note that, although 10X-like data tends to have roughly 1% per
1000 cells captured, the determining factor for doublet formation is the
number of cells inserted into the machine. If for some reason your
recovery rate is lower than expected, you might have a higher doublet
rate than you’d expect from the captured and called cells (in other
words, it would be preferable to say that the doublet rate is roughly
0.6% per 1000 cells put into the machine, where 0.6 is the recovery
rate). In such circumstances, scDblFinder typically sets
the thresholds correctly nevertheless. This is because the thresholding
tries to minimize both the deviation from the expected number of
doublets and the misclassification (i.e. of artificial doublets),
meaning that the effective (i.e. final) doublet rate will differ from
the given one. scDblFinder also considers false positives
to be less problematic than false negatives. You can reduce to some
degree the deviation from the input doublet rate by setting
dbr.sd=0.
Finally, note that version (1.20.0) initially shipped with the
current Bioconductor release (3.20) version included a wrong default
doublet rate (dbr.per1k) argument (it was 0.08 instead of
0.008). This was subsequently fixed in version 1.20.2, but if you
installed before that you might need to update the package.
Should I use the cluster-based doublet generation or not?
Both approaches perform very similarly overall in benchmarks (see the
scDblFinder
paper). If your data is very clearly segregated into clusters, or if
you are interested in the origin of the doublets, the cluster-based
approach is preferable. This will also enable a more accurate accounting
of homotypic doublets, and therefore a slightly better thresholding.
Otherwise, and especially if your data does not segregate very clearly
into clusters, the random approach (e.g. clusters=FALSE,
the default) is preferable.
The clusters don’t make any sense!
If you ran scDblFinder on a multi-sample dataset and did
not provide the cluster labels, then the labels are sample-specific
(meaning that label ‘1’ in one sample might have nothing to do with
label ‘1’ in another), and plotting them on a tSNE will look like they
do not make sense. For this reason, when running multiple samples we
recommend to first cluster all samples together (for example using
sce$cluster <- fastcluster(sce)) and then provide the
clusters to scDblFinder.
‘Size factors should be positive’ error
You will get this error if you have some cells that have zero reads (or a very low read count, leading to zero after feature selection). After filtering out these cells the error should go away.
Identifying homotypic doublets
Like other similar tools, scDblFinder focuses on identifying heterotypic doublets (formed by different cell types), and has only a low performance in identifying homotypic doublets (see this preprint). This can lead to disagreements with doublets called using cell hashes or SNPs in multiplexed samples, which capture both types of doublets similarly (and can miss intra-sample heterotypic doublets, especially if the multiplexing is low). This is why we treat these approaches as complementary.
However, should you for some reason try to identify also homotypic
doublets with scDblFinder, be sure to not to use the cluster-based
approach, and to set removeUnidentifiable=FALSE. Otherwise,
scDblFinder removes artificial doublets likely to be homotypic from
training, therefore focusing the task on heterotypic doublets, but at
the expense ot homotypic ones (which are typically deemed relatively
harmless).
What is a sample exactly? Usage with barcoded and 10X Flex data.
As indicated above, the samples argument should be used
to indicate different captures. For multiplexed samples, this is
expected to be the batch of cells processed together, rather than the
actual samples.
In highly multiplexed datasets such as produced by the 10X Flex kit
(especially 16-plex), this can cause two kinds of problems. First, the
whole logic of the Flex approach is that inter-sample doublets can be
resolved into separate cells, and while a large number of unresolvable
intra-sample doublets will remain (see Howitt
et al., 2024), the expected remaining doublet rate will not be the
same as for classical 10X experiment. For this reason, we recommend to
set a higher dbr.sd in such circumstances,
e.g. dbr.sd=1 to base the thresholding entirely on the
classification accuracy.
Another, more practical problem is that, with such kits, the very
large number of cells in a single capture might translante into very
large computational demands when running scDblFinder. To
circumvent such problem, one can split a batch of cells into more
decently-sized chunks and process the chunks separately, so long as each
chunk is representative of the whole batch in terms of cell
heterogeneity.
How can I make this reproducible?
Because it relies on the partly random generation of artificial
doublets, running scDblFinder multiple times on the same data will yield
slightly different results. You can ensure reproducibility using
set.seed(), however this will not be sufficient when
processing multiple samples (i.e. using the samples
argument – even without multithreading!). In such case, the seed needs
to be passed to the BPPARAMs:
bp <- MulticoreParam(3, RNGseed=1234)
sce <- scDblFinder(sce, clusters="cluster", samples="sample", BPPARAM=bp)Similarly, when processing the samples serially, use
SerialParam(RNGseed = seed).
(Note that in BiocParallel versions <1.28, one had in
addition to explicitly start the cluster before the run using
bpstart(bp), and then bpstop(bp) after
scDblFinder.)
As a final note: when running scDblFinder twice on the
same data with different random seeds, the scores will be highly
correlated, but some cells will be called as doublets (with a high
score) in only one of the runs (e.g. see this issue).
There are good reasons to believe that these are
homotypic doublets (if doublets at all), and if you worry
chiefly about hetertypic doublets, you may concentrate on those that are
reprocibly called across runs.
Can I use this in combination with Seurat or other tools?
If the input SCE already contains a logcounts assay or a
reducedDim slot named ‘PCA’, scDblFinder will use them for
the clustering step. In addition, a clustering can be manually given
using the clusters argument of scDblFinder().
In this way, seurat
clustering could for instance be used to create the artificial doublets
(see ?Seurat::as.SingleCellExperiment.Seurat for conversion
to SCE). For example, assuming as Seurat object
se, the following could be done:
sce <- scDblFinder(GetAssayData(se, slot="counts"), clusters=Idents(se))
# port the resulting scores back to the Seurat object:
se$scDblFinder.score <- sce$scDblFinder.scoreAfter artificial doublets generation, the counts of real and
artificial cells must then be reprocessed (i.e. normalization and PCA)
together, which is performed internally using scater. If
you wish this step to be performed differently, you may provide your own
function for doing so (see the processing argument in
?scDblFinder). We note, however, that the impact of
variations of this step on doublet detection is rather mild. In fact,
not performing any normalization at all for instance decreases doublet
identification accuracy, but by rather little.
For example, the following code would enable the internal use of sctransform:
# assuming `x` is the count matrix:
nfeatures <- 1000
sce <- SingleCellExperiment(list(counts=x))
# sctransform on real cells:
vst1 <- sctransform::vst(counts(sce), n_cells=min(ncol(sce),5000), verbosity=0)
sce <- sce[row.names(vst1$y),]
logcounts(sce) <- vst1$y
hvg <- row.names(sce)[head(order(vst1$gene_attr$residual_variance, decreasing=TRUE), nfeatures)]
# define a processing function that scDblFinder will use on the real+artificial doublets;
# the input should be a count matrix and the number of dimensions, and the output a PCA matrix
myfun <- function(e, dims){
# we use the thetas calculated from the first vst on real cells
e <- e[intersect(row.names(e), row.names(vst1$model_pars_fit[which(!is.na(vst1$model_pars_fit[,"theta"])),])),]
vst2 <- sctransform::vst(e, n_cells=min(ncol(e),5000), method="nb_theta_given",
theta_given=vst1$model_pars_fit[row.names(e),"theta"],
min_cells=1L, verbosity=0)
scater::calculatePCA(vst2$y, ncomponents=dims)
}
sce <- scDblFinder(sce, processing=myfun, nfeatures=hvg)Note however that this did not generally lead to improved performance – but rather decreased on most benchmark datasets, in fact (see comparison in this issue).
{kind=link}
How can I call scDblFinder from the command line?
Here would be an example of how to call scDblFinder (in cluster mode) from the command line and save the results to a csv:
Rscript -e '
library(scDblFinder)
set.seed(123) # for reproducibility
e <- Matrix::readMM("matrix.mtx.gz")
colnames(e) <- readLines("barcodes.tsv.gz")
res <- scDblFinder(e, cluster=TRUE)
res <- cbind(barcode=colnames(res),
colData(res)[,grep("scDblFinder",colnames(colData(res)))])
write.table(res, "output.csv", row.names=FALSE, quote=FALSE)
'Can this be used with scATACseq data?
Yes, see the scATAC vignette specifically on this topic.
Should I run QC cell filtering before or after doublet detection?
The input to scDblFinder should not include empty
droplets, and it might be necessary to remove cells with a very low
coverage (e.g. <200 or 500 reads) to avoid errors. Further quality
filtering should be performed downstream of doublet detection,
for two reasons: 1. the default expected doublet rate is calculated on
the basis of the cells given, and if you excluded a lot of cells as low
quality, scDblFinder might think that the doublet rate
should be lower than it is. 2. kicking out all low quality cells first
might hamper our ability to detect doublets that are formed by the
combination of a good quality cell with a low-quality one. This being
said, these are mostly theoretical grounds, and unless your QC filtering
is very stringent (and it shouldn’t be!), it’s unlikely to make a big
difference.
What about ambiant RNA decontamination?
Contamination by ambiant RNA has emerged as an important confounder in single-cell (and especially single-nuclei) RNAseq data, which prompts the question of whether that should be run prior or after doublet detection. Unfortunately, we do not currently have good evidence pointing in either direction, and arguments can be made for both. Low-quality doublets, or doublets from an experiment with a large dominant celltype, can easily look like contamination, and likewise a high amount of contamination can easily look like a doublet because it includes RNA from other cell types. There is a possibility that a decontamination package sees an actual doublet as contamination, and attempts to clean it, which it will necessarily do imperfectly (because while the decontamination is a mixture of all cells, a doublet isn’t), but perhaps sufficiently so that it can’t be accurately detected as a doublet anymore. This would therefore be an argument for running doublet calling first. However, it’s also possible that decontamination, because it makes the cells cleaner, makes the doublet detection task easier.
Can I combine this method with others?
Of course it is always possible to run multiple methods and combine the results. In our benchmark, the combination of scDblFinder with DoubletFinder, for instance, did yield an improvement in most (though not all) datasets (see the results here), although of a small magnitude. The simplest way is to do an average of the scores (assuming that the scores are on a similar scale, and that a higher score has the same interpretation across methods), which for instance gave similar results to using a Fisher p-value combination on 1-score (interpreted as a probability).
Session information
## R version 4.6.0 (2026-04-24)
## Platform: x86_64-pc-linux-gnu
## Running under: Ubuntu 24.04.4 LTS
##
## Matrix products: default
## BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
## LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
##
## locale:
## [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
## [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
## [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
## [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
## [9] LC_ADDRESS=C LC_TELEPHONE=C
## [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
##
## time zone: Etc/UTC
## tzcode source: system (glibc)
##
## attached base packages:
## [1] stats4 stats graphics grDevices utils datasets methods
## [8] base
##
## other attached packages:
## [1] scDblFinder_1.27.6 SingleCellExperiment_1.35.1
## [3] SummarizedExperiment_1.43.0 Biobase_2.73.1
## [5] GenomicRanges_1.65.0 Seqinfo_1.3.0
## [7] IRanges_2.47.2 S4Vectors_0.51.3
## [9] BiocGenerics_0.59.7 generics_0.1.4
## [11] MatrixGenerics_1.25.0 matrixStats_1.5.0
## [13] BiocStyle_2.41.0
##
## loaded via a namespace (and not attached):
## [1] tidyselect_1.2.1 viridisLite_0.4.3 vipor_0.4.7
## [4] dplyr_1.2.1 farver_2.1.2 viridis_0.6.5
## [7] S7_0.2.2 Biostrings_2.81.3 bitops_1.0-9
## [10] fastmap_1.2.0 RCurl_1.98-1.19 scrapper_1.7.3
## [13] bluster_1.23.0 GenomicAlignments_1.49.0 XML_3.99-0.23
## [16] digest_0.6.39 rsvd_1.0.5 lifecycle_1.0.5
## [19] cluster_2.1.8.2 magrittr_2.0.5 compiler_4.6.0
## [22] rlang_1.2.0 sass_0.4.10 tools_4.6.0
## [25] igraph_2.3.2 yaml_2.3.12 data.table_1.18.4
## [28] rtracklayer_1.73.0 knitr_1.51 S4Arrays_1.13.0
## [31] htmlwidgets_1.6.4 xgboost_3.2.1.1 curl_7.1.0
## [34] DelayedArray_0.39.3 RColorBrewer_1.1-3 abind_1.4-8
## [37] BiocParallel_1.47.0 desc_1.4.3 grid_4.6.0
## [40] beachmat_2.29.0 ggplot2_4.0.3 scales_1.4.0
## [43] MASS_7.3-65 cli_3.6.6 rmarkdown_2.31
## [46] crayon_1.5.3 ragg_1.5.2 otel_0.2.0
## [49] httr_1.4.8 rjson_0.2.23 BiocBaseUtils_1.15.1
## [52] scuttle_1.23.1 ggbeeswarm_0.7.3 cachem_1.1.0
## [55] parallel_4.6.0 BiocManager_1.30.27 XVector_0.53.0
## [58] restfulr_0.0.17 vctrs_0.7.3 Matrix_1.7-5
## [61] jsonlite_2.0.0 bookdown_0.46 BiocSingular_1.29.0
## [64] BiocNeighbors_2.7.2 ggrepel_0.9.8 beeswarm_0.4.0
## [67] irlba_2.3.7 scater_1.41.1 systemfonts_1.3.2
## [70] jquerylib_0.1.4 glue_1.8.1 pkgdown_2.2.0
## [73] codetools_0.2-20 gtable_0.3.6 GenomeInfoDb_1.49.1
## [76] BiocIO_1.23.3 UCSC.utils_1.9.0 ScaledMatrix_1.21.0
## [79] tibble_3.3.1 pillar_1.11.1 htmltools_0.5.9
## [82] R6_2.6.1 textshaping_1.0.5 evaluate_1.0.5
## [85] lattice_0.22-9 Rsamtools_2.29.0 cigarillo_1.3.0
## [88] bslib_0.11.0 Rcpp_1.1.1-1.1 gridExtra_2.3
## [91] SparseArray_1.13.2 xfun_0.58 fs_2.1.0
## [94] pkgconfig_2.0.3